
Diagnostik bei Verdacht auf Morbus Fabry
Wann sollten Fachärzt:innen an Morbus Fabry denken?
Es gibt keine „eindeutigen Morbus Fabry-Symptome“, allerdings sollten bestimmte Symptome, vor allem in Kombination, Ärzt:innen an Morbus Fabry denken lassen und einen Test der Enzymaktivität und ggf. eine genetische Analyse veranlassen:
Direkte Messung der Enzymaktivität im Blutplasma
Hierzu wird Blut des Patienten auf Filterpapier getropft und getrocknet. Die fluorimetrische Messung kann bis zu 6 Monate später erfolgen, da das Enzym stabil ist.
Die Enzymatik in Leukozyten
anhand von EDTA-Blut liefert zuverlässige Ergebnisse.
Messung des Lyso-Gb3-Plasmawertes
Eine verminderte Aktivität der α-GAL A führt zu einem Anstieg des Globotriaosylsphingosinspiegels und hohen Lyso-Gb3-Werten. Normal ist ein Wert ≤ 0,6nmol/l.
Fallen diese Tests positiv aus, gilt eine Morbus Fabry-Diagnose als bestätigt, allerdings schließt ein negatives Testergebnis Morbus Fabry nicht aus. Während Männer mit klassischem Phänotyp (mit wenig bis keiner α-Gal-A-Aktivität) Lyso-Gb3-Normalwerte im Mittel um das 20-Fache übersteigen, haben Männer mit late-onset (mit residueller α-Gal-A-Aktivität) oft Werte nur knapp über den Normwerten. Frauen können sogar im Normalbereich liegen.3
Abklärung des Verdachts auf Morbus Fabry durch einen einfachen Labortest

Eine genetische Analyse des GLA-Gens dient zur Sicherung der Diagnose bei Männern und Frauen und umfasst zumeist eine Mutationsanalyse zur krankheitsauslösenden Mutation, die wiederum wichtige Information für die Wahl der optimalen Therapie liefert.
Fehldiagnosen bei Morbus Fabry: ein breites Spektrum
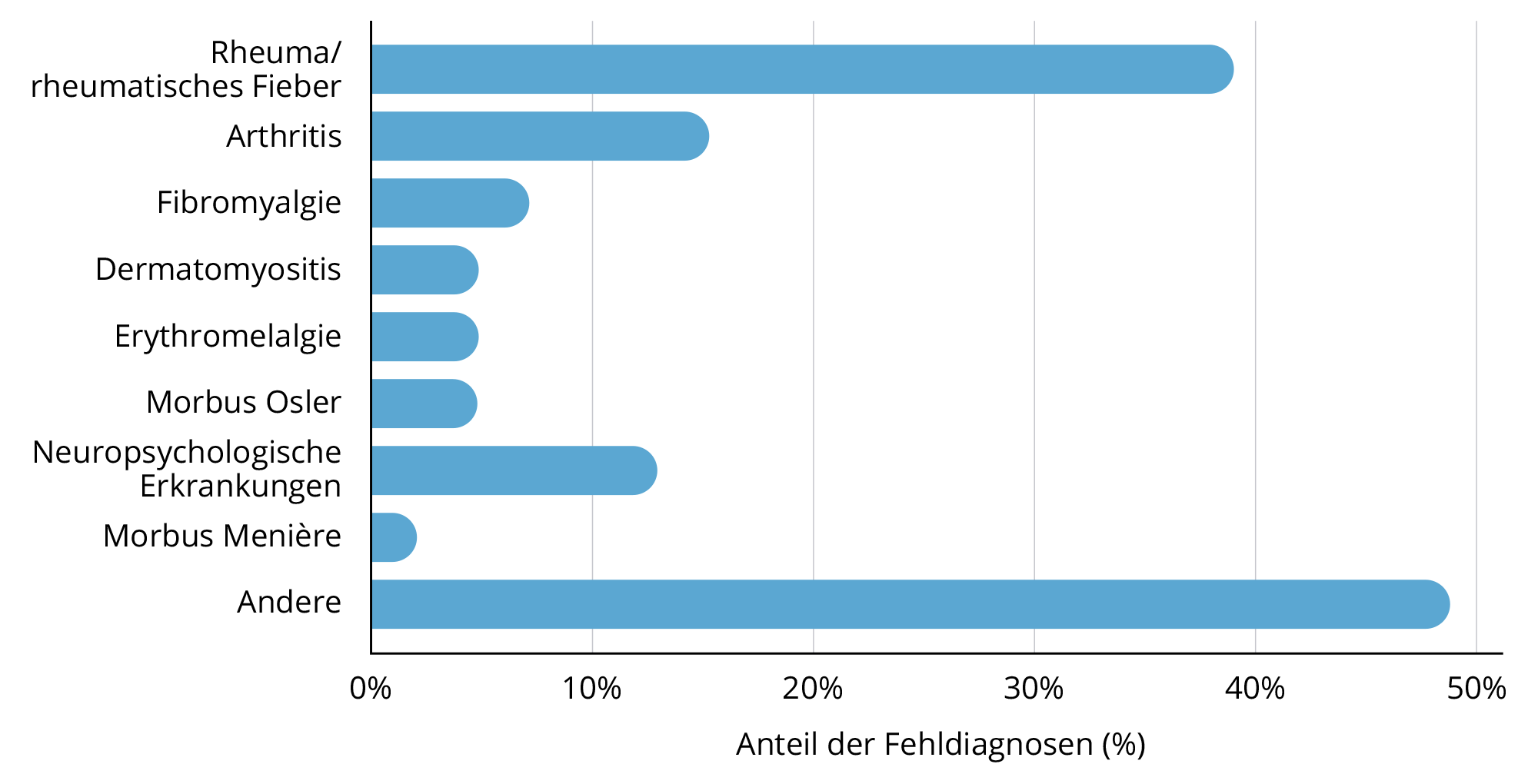
Abb. modifiziert nach METHA, A. et al. 2004
Diagnostik, Symptome
Schubweise auftretende neurologische Symptome
Funktionsstörungen
Lumbalpunktion
Pleozytose (fakultativ)
Oligoklonale Banden*
MRZ-Reaktion positiv**
Andere DD ausgeschlossen
Evozierte Potentiale mit verlängerten Latenzen (MEP, SEP, VEP)
Kraniales MRT
Spinales MRT
KM-Aufnahme örtlicher und zeitlicher Dissemination
Typische Verteilungsmuster der Entmarkungsherde (Bestandteil diagnostischer Kriterien)
Morbus Fabry
-
+
+
+
-
-
+
-
+
-
-
-
Multiple Sklerose
+
+
+
+
+
+
+
+
+
+
+
+
MEP: Magnetisch evozierte Potentiale. MRZ: Masern, Roteln, Zoster. MRT: Magnetresonanztomographie. SEP: Somatosensorisch evozierte Potentiale. VEP: Visuell evozierte Potenziale.
*Sensitivität: 95–100%, **Sensitivität: 90%
Weitere Fehldiagnosen
Mit freundlicher Genehmigung von Prof. Dr. Arndt Rolfs, Universität Rostock
- White Matter Lesions, also Schädigungen der weißen Hirnsubstanz, und damit einhergehende Funktionseinschränkungen treten bei beiden Erkrankungen auf.
- Zöliakie muss bei vorrangig gastrointestinalem Phänotyp ausgeschlossen werden.
- Polyneuropathien werden zwar erkannt, aber die Ursache bleibt oft unklar.
- Bei Kindern und Erwachsenen werden häufig Erkrankungen aus dem rheumatischen Formenkreis diagnostiziert.
- Bei Kindern werden die typischen neuropathischen Schmerzen immer wieder als Wachstumsschmerzen abgetan.



 Kardiologischen Untersuchungsverfahren
Kardiologischen Untersuchungsverfahren
Zu den kardiologischen Untersuchungsverfahren zählen bildgebende Verfahren wie die Elektrokardiographie (EKG), die Echokardiographie (Echo) und die Magnetresonanztomographie (Kardio-MRT mit Late Enhancement Imaging).
Typisch bei Morbus Fabry ist eine linksventrikuläre Hypertrophie zumeist ohne Hypertonie, ein prominenter Papillarmuskel und eine gestörte Klappenfunktion. Im EKG zeigt sich oft eine verkürzte PQ-Zeit und eine T-Wellen-Inversion.8
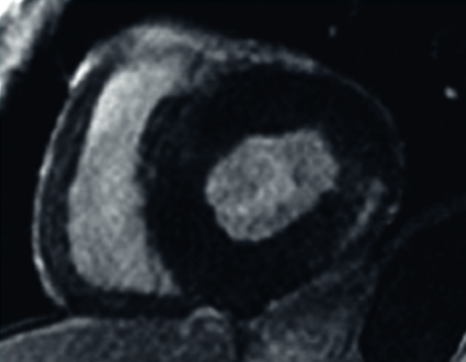
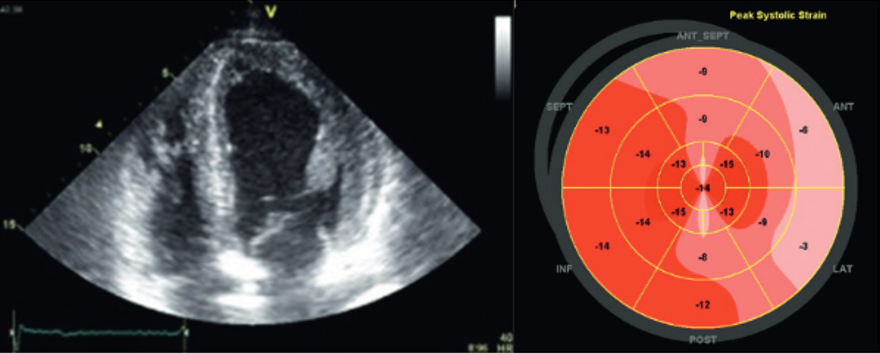
Typische Echokardiographie eines Morbus Fabry-Patienten (links), Magnetresonanztomographie mit Late-Enhancement-Technik zur Darstellung einer myokardialen „Replacement-Fibrose“ (rechts)
Mit freundlicher Genehmigung von Prof. Dr. Frank Weidemann, Recklinghausen
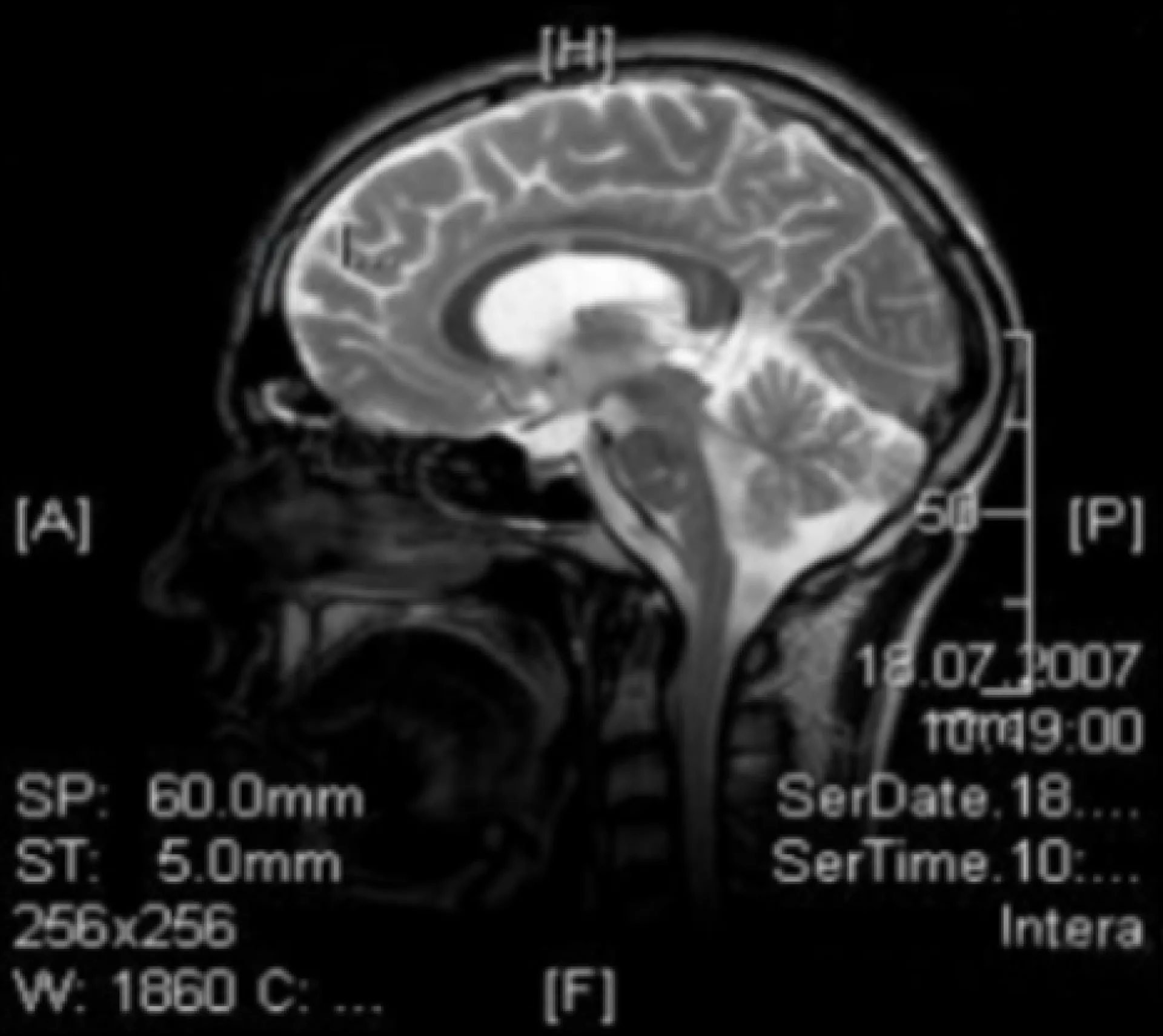
Neurologische Untersuchungen
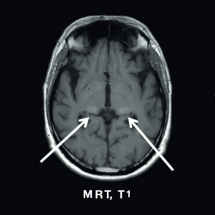
MRT, T1
Bilaterale Signalerhöhung im posterioren Thalamus (“Pulvinar-Zeichen”)
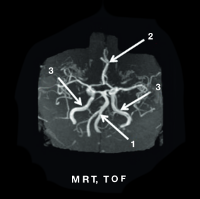
MRT TOF
1. erweiterete A. basilaris
2. A. cerebri anterior
3. A. carotis interna geschlängelte und dilatierte Blutgefäße
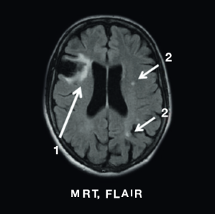
MRT, FLAIR
1. Hirninfarkt cerebri media rechts
2. WML linke Hemispähre
Mit freundlicher Genehmigung von Prof. Dr. Arndt Rolfs, Universität Rostock
Nephrologische Parameter
Nephrologische Parameter sind die Bestimmung des Kreatinins, der Kreatinin-Clearance, der glomerulären Filtrationsrate (GFR) und der Nachweis von Eiweißen im Urin (Mikroalbuminurie bzw. Proteinurie). Nierenbiopsien werden nur empfohlen, wenn der Verdacht besteht, dass es zusätzlich zur Morbus Fabry eine zweite renale Erkrankung vorliegt.
Augen und Ohren
Spaltlampenuntersuchungen zur Diagnose von Cornea verticillata, Tortuositas vasorum und Fabry-Katarakten werden empfohlen. Eine HNO-ärztliche Abklärung kann Hörminderungen, Tinnitus und Störungen im Vestibularapparat aufdecken.
Gastrointestinale Untersuchungen
Gastrointestinale Untersuchungen sind Standarduntersuchungen bei abdominellen Beschwerden und werden nach Bedarf durchgeführt. Infrage kommen ein transabdomineller Ultraschall, eine Gastroskopie oder eine Koloskopie.
Labordiagnostisch
Labordiagnostisch kann der Biomarker Lyso-Gb3 als Erfolgskontrolle für die Therapie verwendet werden.9
Kardiologisch
Die Eingangsuntersuchungen sollten bei Vorliegen kardiologischer Symptome jährlich, ohne Befunde alle 2 Jahre wiederholt werden.
Neurologisch
Neurologische Untersuchungen bei der Diagnosestellung sollten alle 2 Jahre wiederholt werden, falls sie ohne Befund waren. Bei neuauftretenden Symptomen oder Progression ist eine Wiederholung jährlich angezeigt. Dies gilt auch für die Schmerzanamnese und Fragenbögen zur Lebensqualität.
Nephrologisch
Die Eingangsuntersuchungen sollten regelmäßig wiederholt werden. Dazu gehören die Bestimmung des Kreatininwertes, die Kreatinin-Clearance, die glomeruläre Filtrationsrate und die Eiweißausscheidungen im Urin. Sollte sich trotz Therapie eine Progredienz zeigen, kann eine Nierenbiopsie erwogen werden.
Quellen
- REISIN, R. et al. Int J Clin Pract. 2017; 71.
- GERMAIN, D.R. Orphanet J Rare Dis. 2010; 5.30.
- SMID, B.E. et al. J Med Genet. 2015; 52.262–268.
- MEHTA A. et al. Eur J Clin Invest. 2004. 34. 236–242.
- BÖTTCHER, T et al. PLOS ONE. 2013 ; 8.e71894.
- REIBER, H. et al. Mult Scler. 1998; 4(3).111–117.
- HOFFMANN, B. & MAYATEPEK, E. Dtsch Arztebl INT. 2009; 106826.440–447.
- NAMDAR, M. et al. Heart. 2011; 97.485–490.
- NOWAK, A. et al. Mol Genet Metab. 2017; 120.57–61.
- DESNICK, R.J. Prenat Diagn. 2007; 27.693-694.