
Morbus Hunter: Häufige Symptome und mögliche Krankheitsmanifestationen
Der Krankheitsverlauf von Morbus Hunter hängt stark von der Ausprägung ab. Sowohl bei der neuronopathischen, als auch bei der nicht-neuronopathischen Form treten Symptome auf, die mehrere Bereiche des Körpers betreffen, aber nur bei der neuronopathischen Variante hat die Erkrankung Auswirkungen auf das Nervensystem, das Verhalten und die Entwicklung der Betroffenen. Die verschiedenen Symptome, Komplikationen und Komorbiditäten (Begleiterkrankungen) des Hunter-Syndroms erschweren in vielen Fällen eine frühzeitige Diagnose. Meist überschneiden sich die Symptome außerdem mit üblichen „Kinderleiden“ und weisen erst in der Kombination, wie z.B. vermehrte Ohrentzündungen mit skelettalen Veränderungen, auf M. Hunter hin.1,2
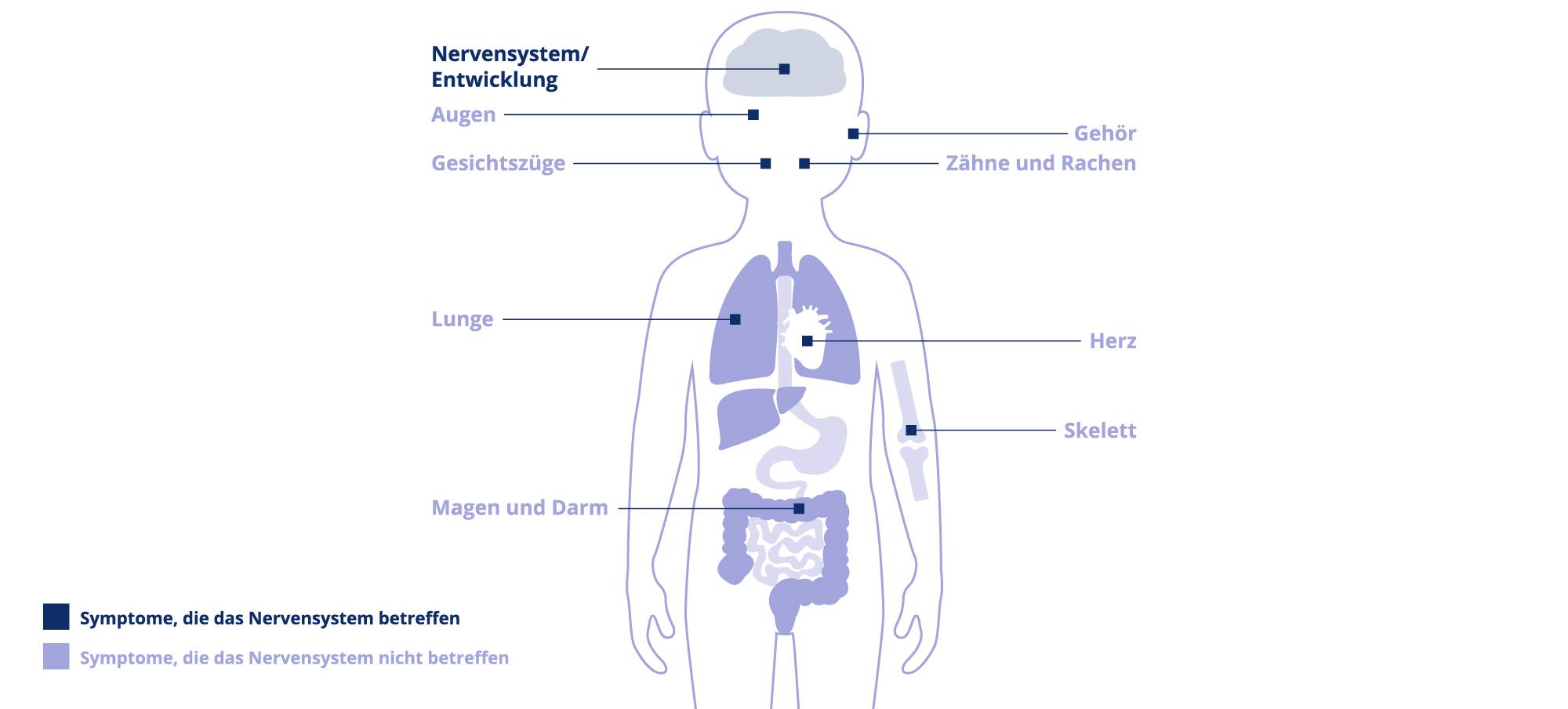



 Schädel und Gesicht
Schädel und Gesicht
- Schädel – Der Schädel ist bei M. Hunter meist vergrößert und die Schädelknochen verdickt. Es kann auch zur Ausprägung eines Hydrocephalus (Wasserkopfs) kommen.
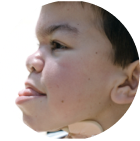
- Gesicht – Dysmorphien (Veränderungen) im Gesicht äußern sich durch vergröberte Gesichtszüge und eine vorstehende Stirn. Die Haare und Augenbrauen Betroffener sind meist sehr struppig und auf der Haut können sich Verdickungen in Form blasser Knötchen bilden, deren Erscheinungsbild auch als Peau d´Orange (Orangenhaut) bezeichnet wird.
- Augen – Bei M. Hunter treten vermehrt Sehstörungen auf, die z. B. durch Veränderungen der Netzhaut oder eine Optikusatrophie (degenerative Sehnerverkrankung) verursacht werden. Das Sehvermögen ist somit eingeschränkt.
- Mundbereich – Die Lippen der Patient:innen sind häufig vergrößert und wirken recht fleischig. Zusätzlich kann eine Makroglossie (vergrößerte Zunge) auftreten. Typisch sind außerdem Prognathie (vorstehender, dominanter Unterkiefer) und ein minderwertiger Zahnschmelz, was eine intensive Zahnpflege notwendig macht.
- Hals – M. Hunter führt auch zu gewissen Einschränkungen im HNO-Bereich (Hals-Nasen-Ohren-Bereich). Der Hals ist meist verkürzt und Stimmveränderungen in Form einer tiefen oder heiseren Stimme können auftreten. Es kann zu einer Häufung von Halsentzündungen mit vergrößerten Mandeln kommen. Die Betroffenen leiden vermehrt an Husten und Dysphagie (Schluckstörungen).
- Ohren – Auch das Innenohr kann betroffen sein: Es kommt zu Gehörstörungen durch häufige Ohrentzündungen, Schwerhörigkeit oder einen kompletten Hörverlust.

Nasen und Atemwege
- Nase – Die Nase der Patient:innen mit M. Hunter ist meist charakteristisch breit und flach geformt. Sie müssen häufig mit Verstopfungen der Nase und Verkühlung kämpfen.
- Atemwege – M. Hunter kann verschiedene Einschränkungen der oberen und unteren Atemwege verursachen. Eingelagerte Mukopolysaccharide (klebrige Kohlenhydrate, die vom Körper gebildet und angesammelt werden) können zu Schleimbildungen im Lungengewebe führen. Häufig treten vermehrt Infekte der Atemwege und allgemeine Atemwegsprobleme wie Atemrhythmusstörungen oder Dyspnoe (Atemnot) auf. Die Störung der Lungenfunktion wirkt sich auch auf den Schlaf aus. Betroffene erleiden oft Schlafapnoen (vorübergehende Atemaussetzer, z.B. durch Blockierung der Atemwege), Schnarchen und einen unruhigen Schlaf.

Bewegungsapparat
- Skelett – Bezeichnend für M. Hunter sind skelettale Veränderungen und Fehlbildungen, die beispielsweise Brustkorb, Wirbelsäule oder Becken betreffen können. Es kann zu Dysostosis multiplex (Knochenfehlbildung) kommen. Die Betroffenen zeigen meist Wachstumsverzögerungen, erreichen eine nur geringe Körpergröße oder sind kleinwüchsig.
- Gang – Durch Fehlstellungen der Hüfte und verkürzte Hüft- bzw. Kniegelenke kann es zu Gangstörungen kommen. Progrediente Gelenkkontrakturen (fortschreitende Einschränkung der Gelenke) verschlimmern das Problem und tragen zusätzlich zur Unbeweglichkeit bei. Außerdem haben Betroffene häufig breite Füße und verkürzte Achillessehnen.
- Hände – Patient:innen mit M. Hunter haben meist kleine und verkürzte Handknochen. Die Hände verformen sich zu sogenannten Klauenhänden. Es kommt vermehrt zum Karpaltunnelsyndrom (Einklemmung der Mittelhandnerven) und zu einer allgemeinen Störung der Fein- und Grobmotorik.
Herz und weitere Organe
- Herz – Bei M. Hunter treten oft kardiologische Probleme wie ein Herzklappenfehler oder eine Kardiomyopathie (Erkrankung des Herzmuskels) auf. Die Herzscheidewand kann undicht sein, was zusätzlich zu einer Herzinsuffizienz führen kann. Eine häufige Todesursache bei M. Hunter-Patient:innen ist das Herzversagen.
- Weitere Organe – Es kann zu Hepatosplenomegalie (Leber- und Milzvergrößerung) und einem allgemein vergrößerten Abdomen (Unterleib) kommen. Außerdem treten vermehrt Schäden an der Niere auf und im Bereich der Eingeweide ist das Risiko für Hernien (Nabel- oder Leistenbrüche) erhöht.
Nervensystem und Verhalten
- Zentrales Nervensystem (nur bei neuronopathischer Form) – Störungen der Sprachfähigkeit und eine verminderte sprachliche Entwicklung sind keine Seltenheit bei M. Hunter. Auch kognitive Fähigkeiten und die allgemeine Lernfähigkeit sind im Vergleich zu gesunden Menschen reduziert. Neben der körperlichen ist somit auch die geistige Entwicklung verzögert, wobei auch eine geistige Rückentwicklung auftreten kann. Je nach Ausprägung der Erkrankung kann die Intelligenz der Patient:innen betroffen sein. Während bei der nicht-neuronopathischen Form meist eine normale Intelligenz vorliegt, reicht das Spektrum bei neuronopathischem M. Hunter von verminderter Intelligenz bis hin zu einer schweren Demenz.
- Verhalten (nur bei neuronopathischer Form) – Patient:innen zeigen häufig Verhaltensauffälligkeiten wie z. B. Hyperaktivität. Auch Lähmungen wie Tetraspastik (Lähmung aller Extremitäten) oder Epilepsie sind typische Symptome der neuronopathischen Form der Erkrankung.

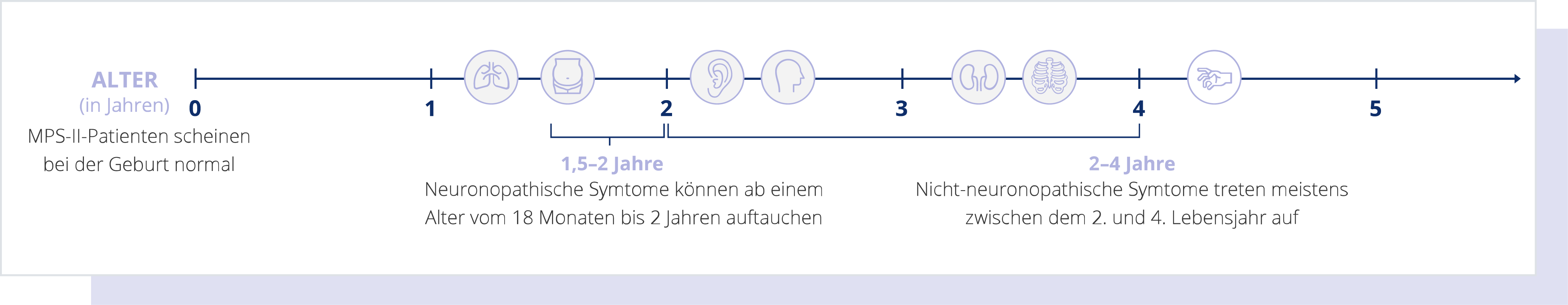
Durch die progrediente Natur der Erkrankung ist eine frühe Diagnose besonders essentiell, sodass so schnell wie möglich mit der Behandlung begonnen werden kann. Bleibt die Erkrankung unbehandelt, ist das Risiko für einen frühzeitigen Tod erhöht. Es ist allerdings wichtig, daran zu denken, dass jedes Kind mit M. Hunter anders ist und es keinen typischen Krankheitsverlauf gibt. Es konnten aber gewisse Unterschiede in der Verlaufsform des neuronopathischen und nicht-neuronopathischen Typs der Erkrankung festgestellt werden.
M. Hunter (MPS II): Neuronopathische Verlaufsform
Beim neuronopathischen Typ von M. Hunter ist zusätzlich das zentrale Nervensystem der Patient:innen betroffen, was sich meist auch auf ihr Verhalten auswirkt. Es kann zu einem Verfall ihrer Sprachfähigkeit, Lern- und anderen kognitiven Fähigkeiten kommen. Die ersten Anzeichen bei neuronopathischem M. Hunter äußern sich meist im Alter von 18 Monaten bis 2 Jahren, was sich z. B. durch Entwicklungsverzögerungen bemerkbar macht.4-7 Zwischen dem 3. und 5. Lebensjahr stagniert die Entwicklung der erkrankten Kinder oft und es treten vermehrt Verhaltensauffälligkeiten wie Hyperaktivität auf. Die neuronopathische Verlaufsform ist die häufigere Ausprägungsform von M. Hunter und betrifft etwa 75% der MPS-II-Patient:innen.5,10
M. Hunter (MPS II): Nicht-neuronopathische Verlaufsform
Die Symptome des nicht-neuronopathischen Typs der Erkrankung treten üblicherweise zwischen dem 2. und 4. Lebensjahr auf.4-7
Mit zunehmendem Alter schreitet die Ausprägung von M. Hunter dann langsamer fort als bei der neuronopathischen Form. Meist verfügen die Patient:innen mit nicht-neuronopathischem M. Hunter über eine normale Intelligenz.2,10
Quellen
1. Martin R et al. Pediatrics 2008;121(2): e377-e386.
2. Gajula P et al. Journal of Natural Science, Biology, and Medicine 2012;3(1): 97.
3. Wraith J E et al. European Journal of Pediatrics 2008;167(3): 267-277.
4. Muenzer J. Rheumatology 2011; 50: v4-v12.
5. Scarpa M et al. Orphanet Journal of Rare Diseases 2011;6(1): 1-18.
6. 1. Wraith J E et al. European Journal of Pediatrics 2008;167(3): 267-277.
7. 1. Burton B K and Giugliani R. European Journal of Pediatrics 2012;171(4): 631-639.
8. Eisengart J B et al. Molecular Genetics and Metabolism Reports 2020;22: 100549.
9. Quaio C R D C et al. JIMD Reports-Case and Research Reports, 2012/1, 2011: 125-128.
10. Downs A T, Crisp D T and Ferretti D G. Pediatric Dentistry 1995;17(2): 98-100.