
Morbus Fabry: Symptome und mögliche Krankheitsmanifestationen
Es existieren über 1.000 verschiedene Genmutationen, die für Morbus Fabry verantwortlich sind, und je nach Mutation und Restaktivität des Enzyms kann es zu unterschiedlich ausgeprägten Krankheitsmanifestationen und zu einer Vielzahl von individuellen Symptomen kommen. Bei manchen Genmutationen wird keine funktionsfähige Galaktosidase gebildet, entsprechend sind die Symptome stark ausgeprägt und treten früher auf.
Morbus Fabry ist eine multisystemische Erkrankung, das bedeutet, dass mehrere Organsysteme betroffen sind, außerdem kann die Krankheit unterschiedlich stark ausgeprägt sein. Häufig sind folgende Organsysteme betroffen:
Kardio-vaskulär (Herz-Kreislauf)
Das Herz ist besonders oft betroffen bei Morbus Fabry, Veränderungen sind aber meist nur für den Arzt erkennbar. Typisch sind linksventrikuläre Hypertrophie, Arrhythmien und ein prominenter Papillarmuskel.
Nephrologisch (Nieren)
Die Nieren leiden sehr oft bei Morbus Fabry, dies äußert sich als Proteinurie (Eiweiß im Urin), Niereninsuffizienz (Nierenversagen) und parapelvine Zysten (Zysten im Hüftbereich)
Zerebrovaskulär (Gehirn)
Schlaganfall und transitorische ischämische Attacken (ein vorübergehender Schlaganfall) treten schon bei Patient:innen unter 55 Jahren auf, im MRT sind White Matter Lesions zu erkennen. Häufig treten auch Depressionen auf.
Neurologisch (Nervensystem)
Schädigungen von Nervenzellen erzeugen neuropathische Schmerzen, häufig äußern diese sich als brennende Schmerzen in Händen und Füßen.
Gastro (Magen und Darm)
Diese äußern sich als Bauchschmerzen, Diarrhö/Durchfall, Völlegefühl und Übelkeit.
Dermatologisch (Haut)
Angiokeratome sind ein typisches Symptom bei Morbus Fabry, sie bezeichnen einen Ausschlag mit kleinen roten Punkten mit einer verdickten Haut. Hypo- und Anhidrose sind ebenfalls typisch, was bedeutet, dass die Patient:innen zu wenig oder gar nicht schwitzen können.
Die Symptome von Morbus Fabry sind vielfältig und bei jedem/jeder Patient:in individuell unterschiedlich. Außerdem verändern sie sich im Krankheitsverlauf.
Manche Patient:innen leiden unter unspezifischen Symptomen wie Schmerzen, wiederkehrenden Fieberschüben (vor allem bei Kindern), chronischer Fatigue und Depressionen. Die dauerhaften Beschwerden führen oft zu einer reduzierten körperlichen Belastbarkeit und einer eingeschränkten Lebensqualität. Typisch sind sogenannte Fabry-Krisen, dabei handelt es sich um Schmerzphasen, die ein paar Minuten oder auch mehrere Tage andauern können.
Die Symptome sind bei Männern und Frauen unterschiedlich. Männer haben meist stärkere Symptome mit einem früheren Krankheitsbeginn, während Frauen eine weite Spanne an Krankheitszeichen zeigen können. Diese reichen von kaum Symptomen bis hin zu einen Krankheitsverlauf vergleichbar dem bei Männern. Der Grund dafür ist, dass die Genmutationen, die für Morbus Fabry verantwortlich sind, auf dem X-Chromosom liegen. Frauen haben zwei X-Chromosomen, in der Regel ist nur eines betroffen, sie haben also ein zweites, gesundes X-Chromosom, das zum Teil das Enzymdefizit ausgleichen kann. Männer dagegen besitzen neben einem Y-Chromosom nur ein X-Chromosom.1

Dies führt häufig zu Fehldiagnosen. Ärzt:innen müssen daher alle Symptome auswerten und die Restaktivität des Enzyms alpha-Galaktosidase A testen. Die Höhe der Enzymrestaktivität bestimmt den zukünftigen Krankheitsverlauf sowie die Therapie und kann Hinweise auf die zu erwartenden Symptome geben. Der Verlauf ist zwar insgesamt progredient, also fortschreitend, aber die individuelle Entwicklung ist nicht genau vorhersehbar. Jede Zelle und jedes Gewebe werden von der krankhaften Speicherung der Glykospingolipiden zunehmend geschädigt, daher ist Morbus Fabry eine Multiorganerkrankung. Je nachdem, welche Anzeichen im Vordergrund stehen, kann man eine Herzvariante und eine Nierenvariante unterschieden.
Medianes kumulatives Überleben von unbehandelten Fabry-PatientInnen


Abb. modifiziert nach MACDERMONT, KD. et al. 20012,3
Patient:innen mit unbehandeltem Morbus Fabry haben gegenüber der Allgemeinbevölkerung eine deutlich verkürzte Lebenserwartung. Das mediane Überleben liegt bei Männern bei 49 Jahren und damit etwa 20 Jahre unter der von nicht betroffenen Männern.2 Bei Frauen ist der Unterschied geringer, betroffene Frauen haben eine Lebenserwartung von 70 Jahren und liegen damit etwa 15 Jahre unter der nicht betroffener Frauen.3
Zur Diagnose der Fabry-Krankheit bestimmen Ärzt:innen die Enzymaktivität mithilfe eines Trockenbluttests. Wenn nicht ausreichend Enzymaktivität vorhanden ist, kann der Krankheitsmarker Lyso-Gb3 im Blut nachgewiesen werden. Bei Männern reicht dies zur Diagnosestellung aus, bei Frauen ist eine zusätzliche molekulargenetische Diagnostik notwendig.
Eine Stammbaumanalyse bei dieser Erbkrankheit durchzuführen, ist ebenfalls von großer Bedeutung, um weitere Betroffene, die möglichweise eine starke Symptomatik haben, aber noch nicht diagnostiziert worden sind, zu finden und ihnen damit die Möglichkeit einer Therapie zukommen zu lassen.


Wie selten ist Morbus Fabry?
Morbus Fabry gehört zu den seltenen Erkrankungen.
Was bedeutet das? Schätzungen zufolge leidet einer von 40.000 Männern und eine von 20.000 Frauen4 an Morbus Fabry. Im vollbesetzten Berliner Olympiastadion hätten nur eine oder zwei Personen Morbus Fabry! Jedoch wird eine hohe Dunkelziffer vermutet. Untersuchungen bei Neugeborenen zeigen ein wesentlich höheres Vorkommen von 1:1.250 bis 1:4.600.5,7

Mediales kumulutatives Überleben

Abb. modifiziert nach ENG, CM et al. 2007
Da Morbus Fabry eine erblich bedingte und fortschreitende Erkrankung ist, beginnen die Krankheitsmanifestationen bereits während der embryonalen Entwicklung. Durch die gestörten Stoffwechselvorgänge sammeln sich immer mehr Abfallprodukte in den Zellen und verursachen einen progredienten Verlauf. Erste Symptome und Krankheitszeichen treten häufig bereits in der Kindheit auf in Form von brennenden Schmerzen und Kribbeln in den Händen und Füßen. Mit zunehmendem Alter kommen Gewebeschäden hinzu, die unbehandelt schließlich zu Organversagen führen können.
Alter der Fabry-Patienten beim Einsetzen der Symptome

Abb. modifiziert nach CYBULLLA, M. et al. 2007
Ohne Therapie treten progrediente Organschäden auf, die mit zunehmendem Alter stärker werden. Zu den klassischen Leitsymptomen zählen:
Der Wandel im Verlauf des Lebens
Symptome in der Kindheit
- Brennende Schmerzen
- Kribbeln in Händen und Füßen
- Unerklärbare Fieberschübe
- Hornhauttrübung
- Verminderte Fähigkeit zu schwitzen
- Bauchschmerzen und Durchfall
Symptome bei Jugendlichen
- Brennende Schmerzen und Kribbeln in Händen und Füßen
- Verminderte Fähigkeit zu schwitzen
- Verminderte Leistungsfähigkeit, erhöhte Müdigkeit
- Hornhauttrübung
- Rötlich-bläuliche, punktförmige Angiokeratome
- Magen-Darm-Probleme
Symptome bei Erwachsenen
- Verminderte Fähigkeit zu schwitzen
- Hornhauttrübung
- Rötlich-bläuliche, punktförmige Angiokeratome
- Nierenfunktionsstörungen
- Auffälligkeiten am Herzen
- Hörsturz
- Schwindel
- Schlaganfall vor dem 55. Lebensjahr



 Nephrologische Symptome: Beteiligung der Nieren bei Morbus Fabry
Nephrologische Symptome: Beteiligung der Nieren bei Morbus Fabry
Das erste Symptom ist meist eine Mikroalbuminurie, die mildeste Form der Proteinurie. Sie bezeichnet eine Ausscheidung von geringen Mengen Eiweiß im Urin, die bereits in der zweiten Lebensdekade beginnen kann.11 Im späteren Verlauf verstärkt sich die Proteinurie, einhergehend mit einer progredienten Abnahme der GFR (glomeruläre Filtrationsrate) bis hin zur Dialysepflicht.1 Eine Nierentransplantation kann notwendig werden.
Bei Frauen mit Morbus Fabry liegt nicht unbedingt eine Nierenschädigung vor. Da sie zumeist nur ein X-Chromosom mit mutiertem GLA-A-Gen haben, während das andere gesund ist. In jeder Körperzelle wird ein X-Chromosom inaktiviert. Es kann sein, dass in der Niere das gesunde X-Chromosom aktiv ist und funktionsfähige Alpha-Galaktosidase A vorhanden ist. Bei Verdacht auf eine Nierenfunktionsstörung ist eine Nierenbiopsie zur Bestimmung der Enzymaktivität notwendig. Bei Männern liegt in der Regel eine Nierenbeteiligung vor, da sie nur über ein X-Chromosom verfügen.
Progrediente Niereninsuffizienz: eGFR-Verlauf ohne Therapie

Kardiologische Symptome: Beteiligung des Herzens bei Morbus Fabry
Kardiale Manifestationen gehören zu den häufigen Symptomen bei Morbus Fabry, dazu gehören linksventrikuläre Hypertrophie (LVH) zumeist ohne einen Hypertonus, ein verdickter Papillarmuskel, Arrhythmien und eine myokardiale Fibrose.14,15


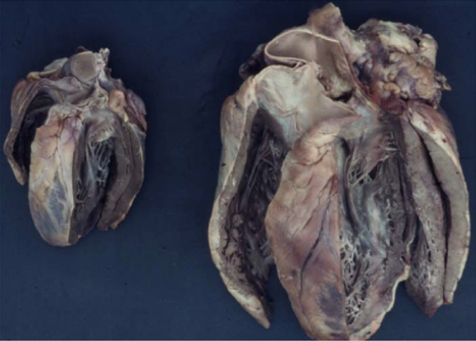
Vergleich eines normalen Herzens und eines hypertrophen Herzens eines Morbus Fabry- Patienten
Eine Beteiligung des Herzens liegt bei etwa 80% der Fabry-Patient:innen vor12, im Alter von durchschnittlich 36 Jahren haben schon über 50% Fabry-Symptome am Herzen.15,17 Diese reichen von Luftnot bei Belastung, Herzrhythmusstörungen, Erkrankungen des Herzmuskels (Kardiomyopathie) bis hin zu Herzklappenfehlern (Klappenanomalien). Durch die Einlagerung von Speichermaterial in die Herzmuskelzellen, Herzklappen und Wände der Blutgefäße am Herzen werden diese geschädigt, sodass es bereits bei Kindern zu Herzinsuffizienz und kardialen Symptomen kommen kann. Im späteren Krankheitsverlauf gibt es ein erhöhtes Risiko für maligne Herzrhythmusstörungen, Herzinsuffizienz und akuten Myokardinfarkt.17
Bei Frauen tritt oft eine Myelofibrose auf, noch bevor es zu einer Vergrößerung des Herzens und damit einhergehenden Herzbeschwerden kommt.18
Im EKG können sich außerdem eine verkürzte PQ-Zeit, eine T-Wellen-Inversion und AV-Blockierungen zeigen.1,19


Schematische Darstellung der Veränderung im EKG bei Morbus Fabry
Neurologische Symptome: Beteiligung des Nervensystems bei Morbus Fabry
Bei Morbus Fabry können sowohl das periphere Nervensystem (PNS) als auch das zentrale Nervensystem (ZNS) betroffen sein. Neurologische Symptome sind eine gestörte Schweißbildung, brennende Schmerzen in Händen und Füßen bis hin zu Durchblutungsstörungen im Gehirn mit frühen transitorischen ischämischen Attacken (TIA) und frühen Schlaganfällen.
Unter neuropathischen Schmerzen oder Nervenschmerzen leidet ein Großteil der Fabry-Patient:innen, insbesondere Männer.20 Typisch sind wiederkehrende Brennschmerzen in den distalen Gliedmaßen (Hände und Füße), aber auch in milderer Form Missempfindungen wie Taubheit, Kribbeln und Ameisenlaufen. Sie entstehen durch Schädigungen der Nervenfasern, auch Polyneuropathien genannt. Die Ursache ist eine Dysfunktion der nicht-myelinisierten C-Fasern, die für das Schmerzempfinden verantwortlich sind.
Die Schmerzlokalisation bei Männern und Frauen mit Morbus Fabry ist unterschiedlich.
Schmerzlokalisation bei 287 Fabry-Patienten (148 Männer, 139 Frauen)

Abb. modifiziert nach HOFFMANN, B. et al. 2007.21
Die Ablagerung von Speicherstoffen in den Schweißdrüsen verursacht eine verminderte oder komplette Unfähigkeit zu schwitzen (Hypo-/Anhidrose), sowie eine Kälte- und Hitzeüberempfindlichkeit. Daher können Temperaturwechsel, körperliche Anstrengung oder auch Fieber Trigger für Fabry-Schmerzen, sogenannte Fabry-Krisen, sein.
Oftmals sind zerebrovaskuläre Symptome wie Schlaganfälle und transitorische ischämische Attacken (TIA) die Erstmanifestationen der Krankheit. So ist das Schlaganfallrisiko gegenüber der Allgemeinbevölkerung erhöht, es wird auf 6,9% für Männer und 4,3% für Frauen geschätzt.1 Schlaganfälle im Alter unter 55 Jahren werden als juvenile Schlaganfälle bezeichnet, sie treten bei Morbus Fabry häufig auf, mit einem medianen Alter von 39 Jahren bei Männern und 46 Jahren bei Frauen. Die Ursache sind vermutlich Mikroangiopathien. Weitere Gefäßerkrankungen im Gehirn sind Mikroangiopathien, intrazerebrale Blutungen, subarachnoidale Blutungen, Mikroblutungen und Hirnvenenthrombosen. Im MRT sind häufig schon bereits Gehirnläsionen, sogenannte White Matter Lesions oder Marklagerläsionen, erkennbar, selbst wenn noch keine Symptome wie Hemiplegien und Hemiparästhesien vorhanden sind.
Gastrointestinale Beschwerden: Beteiligung des Magen-Darm-Traktes bei Morbus Fabry
Magen-Darm-Beschwerden treten bei etwa 50–70% der Fabry-Patient:innen16 auf. Sie gehören zu den frühen Symptomen, so dass bereits Kinder mit M. Fabry unter Bauchschmerzen und abdominellen Krämpfen, vor allem nach dem Essen, leiden. Häufige Durchfälle, Übelkeit und Erbrechen treten ebenfalls häufig auf und können zu einer Unterernährung führen. Es ist aber auch das Gegenteil, also Obstipation (Verstopfung), beschrieben.

Abb. modifiziert nach HOFFMANN, B. et al. 2007.16
Dermatologische Symptome: Beteiligung der Haut bei Morbus Fabry
Das äußerlich auffälligste Symptom von Morbus Fabry sind Angiokeratome in Form eines purpurroten Hautausschlags, vor allem im Bereich von Leisten/Lenden, Oberschenkeln, Gesäß, Schleimhäuten, auf dem Lippenrot und rund um den Bauchnabel (periumbilikal). Sie treten schon früh im Krankheitsverlauf auf.

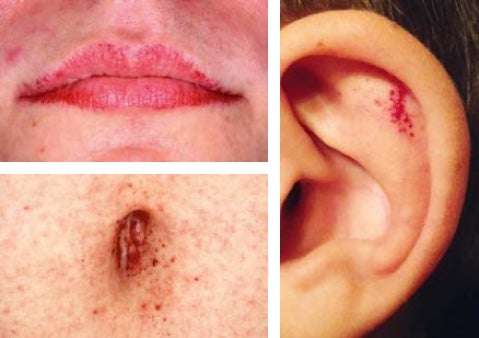
Fotos von Fabry-typischen Angiokeratomen an Mund, Ohr und Bauchnabel
Mit freundlicher Genehmigung von PD Dr. Thomas Jansen, Bochum.
Die Angiokeratome entstehen durch lokale Vermehrung von Blutgefäßen, sodass sie als stecknadelkopfroße rote Punkte auf der Haut sichtbar werden. Ihre Anzahl kann variieren, wobei nicht jede:r Fabry-Patient:in Angiokeratome ausbildet.
Ophthalmologische Symptome: Beteiligung der Augen bei Morbus Fabry

Cornea verticillata

Geschlängelte konjuktivale Gefäße

Geschlängelte retinale Gefäße
Auditorische und vestibuläre Symptome: Beteiligung des Gehörs und des Innenohrs bei Morbus Fabry
Zumeist tritt ein langsamer, progressiver Hörverlust auf. Auditorische und vestibuläre Abnormitäten können außerdem Hörstürze, Tinnitus und Schwindel zur Folge haben.23
Quality-of-Life-Scores von schwer betroffenen Frauen bei Morbus Fabry im Vergleich zur deutschen weiblichen Allgemeinbevölkerung

Abb. modifiziert nach BAEHNER, F. et al. 2003.25
Quellen
- GERMAIN, D.P. Orphanet J Rare Dis. 2010. 5. 30.
- MACDERMOT, K.D. et al. J Med Genet. 2001a. 38(11). 750–760.
- MACDERMOT, K.D. et al. J Med Genet. 2001b. 38(11). 769–775.
- DESNICK, R.J. et al. In: Scriver CR et al. (Hrsg.) The metabolic and molecular bases of inherited disease, 8th ed. New York: McGraw-Hill, 2001. 3733–3774.
- SPADA, M. et al. Am J Hum Genet. 2006. 79. 31–40.
- HWU, W.L. et al. Hum Mutat. 2009. 30. 1397–1405.
- MECHTLER, T.P. et al. Lancet. 2012. 379. 335–341.
- MEHTA, A. et al. Eur J Clinical Invest. 2004. 34(3). 236–42.
- HOPKIN, R.J. et al. Pediatr Res. 2008. 64. 550–5
- SESTITO, S. et al. Curr Pharm Des. 2013. 19. 6037–45
- NAJAFIAN, B. et al. Pediatr Nephrol. 2013. 28. 679–87.
- WHYBRA C et al. Klinische Manifestation des Morbus Fabry bei Kindern. In: Neuropädiatrie 2, 2005. 44–4.
- WEST, M. et al. J Am Soc Nephrol. 2009; 20(5). 1132–1139.
- HOFFMANN, B. et al. Clin Gastroenterol Hepathol. 2007; 5.1447–53.
- WEIDEMANN, F. et al. Int J Cardiol. 2010; 141(1).3–10.
- KAMPANN, C. et al. Orphanet J Rare Dis. 2015. 10. 125.
- LINHART, A. et al. Eur Heart J 2007; 28.1228–35.
- WEIDEMANN, F. et al. Mol Genet Metab. 2019; 126(2).169–82.
- NAMDAR, M. et al. Heart. 2011; 97(6).485–990.
- ÜÇEYLER, N et al. Clin J Pain 2014; 30.915–920.
- HOFFMANN, B. et al. Clin J Pain. 2007 ; 23.535–42.
- SODI, A. Br J Ophthalmol 2007; 91.210–214.
- GERMAIN, D.P. BMC Med Genet 2002; 3.10.
- MINERS, A.H. et al. Qual Life Res 2002 ; 11.127–133.
- BAEHNER, F. et al. J Inherit Metab Dis. 2003; 26(7).617–62.